Quality Assessment with htseq-qa¶
The Python script htseq-qa takes a file with sequencing reads (either
raw or aligned reads) and produces a PDF file with useful plots to assess
the technical quality of a run.
Plot¶
Here is a typical plot:
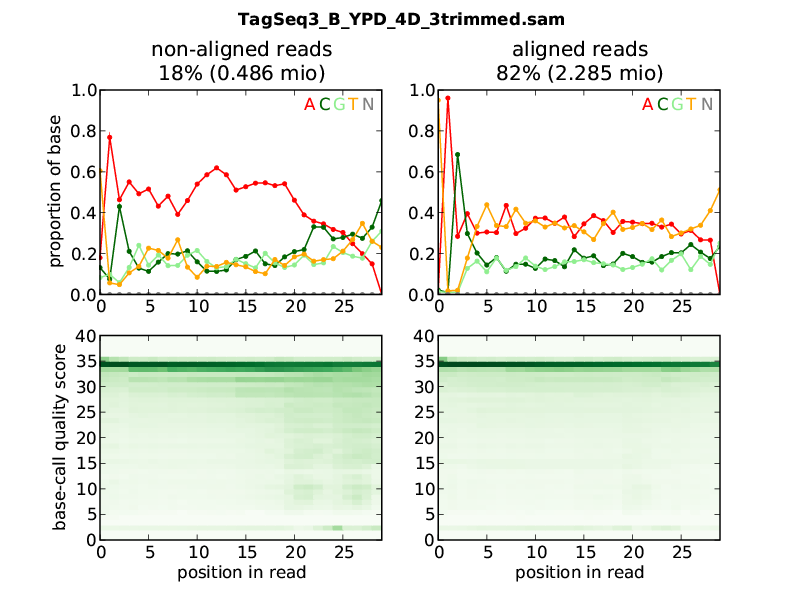
The plot is made from a SAM file, which contained aligned and unalignable reads. The left column is made from the non-aligned, the right column from the aligned reads. The header informs you about the name of the SAM file, and the number of reads.
The upper row shows how often which base was called for each position in the read. In this sample, the non-alignable reads have a clear excess in A. The aligned reads have a balance between complementing reads: A and C (reddish colours) have equal levels, and so do C and G (greenish colours). The sequences seem to be AT rich. Furthermore, nearly all aligned reads start with a T, followed by an A, and then, a C in 70% and an A in 30% of the reads. Such an imbalance would be reason for concern if it has no good explanation. Here, the reason is that the fragmentation of the sample was done by enzyme digestion.
The lower half shows the abundance of base-call quality scores at the different positions in the read. Nearly all aligned reads have a quality of 34 over their whole length, while for the non-aligned reads, some reads have lower quality scores towards their ends.
Usage¶
Note that htseq-qa needs matplotlib to produce the plot, so you need to install this
module, as described here on the matplotlib web site.
After you have installed HTSeq (see Prequisites and installation) and matplotlib, you can run htseq-qa from
the command line:
htseq-qa [options] read_file
If the file htseq-qa is not in your path, you can, alternatively, call the script with
python -m HTSeq.scripts.qa [options] read_file
The read_file is either a FASTQ file or a SAM file. For a SAM file, a plot with two columns is produced as above, for a FASTQ file, you get only one column.
The output is written into a file with the same name as read_file, with the suffix .pdf
added. View it with a PDF viewer such as the Acrobat Reader.
Options¶
-
-t<type>,--type=<type>¶ The file type of the read_file. Supported values for <type> are:
sam: a SAM file (Note that the SAMtools contain Perl scripts to convert most alignment formats to SAM)solexa-export: an_export.txtfile as produced by the SolexaPipeline software after aligning with Eland (htseq-qaexpects the new Solexa quality encoding as produced by version 1.3 or newer of the SolexaPipeline)fastq: a FASTQ file with standard (Sanger or Phred) quality encodingsolexa-fastq: a FASTQ file with Solexa quality encoding, as produced by the SolexaPipeline after base-calling with Bustard (htseq-qaexpects the new Solexa quality encoding as produced by version 1.3 or newer of the SolexaPipeline)
-
-o<outfile>,--outfile=<outfile>¶ output filename (default is <read_file>``.pdf``)
-
-r<readlen>,--readlength=<readlen>¶ the maximum read length (when not specified, the script guesses from the file
-
-g<gamma>,--gamma=<gamma>¶ the gamma factor for the contrast adjustment of the quality score plot
-
-n,--nosplit¶ do not split reads in unaligned and aligned ones, i.e., produce a one-column plot
-
-m,--maxqual¶ the maximum quality score that appears in the data (default: 40)
-
-h,--help¶ Show a usage summary and exit