htseq-count: counting reads within features
Given a file with aligned sequencing reads and a list of genomic features, a common task is to count how many reads map to each feature.
A feature is here an interval (i.e., a range of positions) on a chromosome or a union of such intervals.
In the case of RNA-Seq, the features are typically genes, where each gene is considered here as the union of all its exons. One may also consider each exon as a feature, e.g., in order to check for alternative splicing. For comparative ChIP-Seq, the features might be binding region from a pre-determined list.
Special care must be taken to decide how to deal with reads that align to or
overlap with more than one feature. The htseq-count script allows to
choose between three modes. Of course, if none of these fits your needs,
you can write your own script with HTSeq. See the chapter A tour through HTSeq for a
step-by-step guide on how to do so. See also the FAQ at the end, if the
following explanation seems too technical.
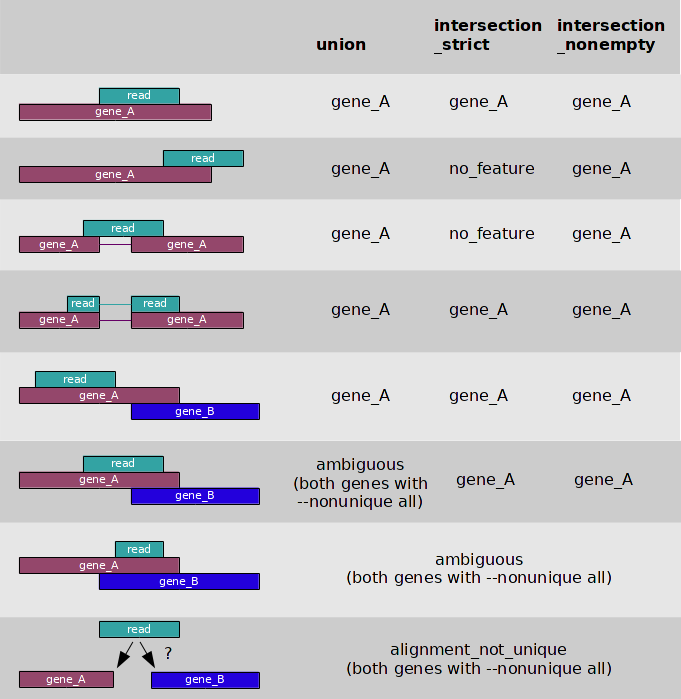
The three overlap resolution modes of htseq-count work as follows. For
each position i in the read, a set S(i) is defined as the set of all
features overlapping position i. Then, consider the set S, which is
(with i running through all position within the read or a read pair)
the union of all the sets S(i) for mode
union. This mode is recommended for most use cases.the intersection of all the sets S(i) for mode
intersection-strict.the intersection of all non-empty sets S(i) for mode
intersection-nonempty.
If S contains precisely one feature, the read (or read pair) is counted for this feature. If
S is empty, the read (or read pair) is counted as no_feature. If S
contains more than one feature, htseq-count behaves differently based on
the --nonunique option:
--nonunique none(default): the read (or read pair) is counted asambiguousand not counted for any features. Also, if the read (or read pair) aligns to more than one location in the reference, it is scored asalignment_not_unique.--nonunique all: the read (or read pair) is counted asambiguousand is also counted in all features to which it was assigned. Also, if the read (or read pair) aligns to more than one location in the reference, it is scored asalignment_not_uniqueand also separately for each location.--nonunique fraction: the read (or read pair) is counted asambiguousand is also counted fractionally in all features to which it was assigned. For example, if the read overlaps with 3 features, it will be counted 1/3 to each of them.--nonunique random: the read (or read pair) is counted asambiguousand is also counted uniformly at random toone ofthe features to which it was assigned.
Notice that when using --nonunique all the sum of all counts will not
be equal to the number of reads (or read pairs), because those with multiple
alignments or overlaps get scored multiple times. By contrast, with
--nonunique fraction or --nonunique random, the sum of all counts
will be equal to the number of reads (or read pairs).
The following figure illustrates the effect of these three modes and the
--nonunique option:
Usage
After you have installed HTSeq (see Installation), you can run htseq-count from
the command line:
htseq-count [options] <alignment_files> <gtf_file>
If the file htseq-count is not in your path, you can, alternatively, call the script with
python -m HTSeq.scripts.count [options] <alignment_files> <gtf_file>
The <alignment_files> are one or more files containing the aligned reads in
SAM/BAM/CRAM format. Under the hood, we use pysam for automatic file type detection,
so whatever pysam can parse we can too (SAMtools can convert most alignment formats
to one of these.) Make sure to use a splicing-aware aligner such as STAR.
htseq-count makes full use of the information in the CIGAR field.
To read from standard input, use - as <alignment_files>.
If you have paired-end data, pay attention to the -r option described below.
The <gtf_file> contains the features in the GTF format.
The script outputs a table with counts for each feature, followed by the special counters, which count reads that were not counted for any feature for various reasons. The names of the special counters all start with a double underscore, to facilitate filtering. (Note: The double unscore was absent up to version 0.5.4). The special counters are:
__no_feature: reads (or read pairs) which could not be assigned to any feature (set S as described above was empty).__ambiguous: reads (or read pairs) which could have been assigned to more than one feature and hence were not counted for any of these, unless the--nonunique alloption was used (set S had more than one element).__too_low_aQual: reads (or read pairs) which were skipped due to the-aoption, see below__not_aligned: reads (or read pairs) in the SAM file without alignment__alignment_not_unique: reads (or read pairs) with more than one reported alignment. These reads are recognized from theNHoptional SAM field tag. (If the aligner does not set this field, multiply aligned reads will be counted multiple times, unless they getv filtered out by due to the-aoption.) Note that if the--nonunique alloption was used, these reads (or read pairs) are still assigned to features.
Important: The default for strandedness is yes. If your RNA-Seq data has not been made
with a strand-specific protocol, this causes half of the reads to be lost.
Hence, make sure to set the option --stranded=no unless you have strand-specific
data!
Important: For paired-end reads, although position-sorted BAM files are supported, unsorted BAM files (i.e. in which the two reads of the pair are in consecutive lines of the BAM file) are highly recommended for htseq-count. If you are having trouble or unexpected results, sort your BAM file by name and try again.
Options
- -f <format>, --format=<format>
Format of the input data. Possible values are
sam(for text SAM files) andbam(for binary BAM files). Default issam.DEPRECATED: Modern versions of samtools/htslibs, which HTSeq uses to access SAM/BAM/CRAM files, have automatic file type detection. This flag will be removed in future versions of htseq-count.
- -r <order>, --order=<order>
For paired-end data, the alignment have to be sorted either by read name or by alignment position. If your data is not sorted, use the
samtools sortfunction ofsamtoolsto sort it. Use this option, withnameorposfor<order>to indicate how the input data has been sorted. The default isname.If
nameis indicated,htseq-countexpects all the alignments for the reads of a given read pair to appear in adjacent records in the input data. Forpos, this is not expected; rather, read alignments whose mate alignment have not yet been seen are kept in a buffer in memory until the mate is found. While, strictly speaking, the latter will also work with unsorted data, sorting ensures that most alignment mates appear close to each other in the data and hence the buffer is much less likely to overflow.
- --max-reads-in-buffer=<number>
When <alignment_file> is paired end sorted by position, allow only so many reads to stay in memory until the mates are found (raising this number will use more memory). Has no effect for single end or paired end sorted by name. (default:
30000000)
- -s <yes/no/reverse>, --stranded=<yes/no/reverse>
Whether the data is from a strand-specific assay (default:
yes)For
stranded=no, a read is considered overlapping with a feature regardless of whether it is mapped to the same or the opposite strand as the feature. Forstranded=yesand single-end reads, the read has to be mapped to the same strand as the feature. For paired-end reads, the first read has to be on the same strand and the second read on the opposite strand. Forstranded=reverse, these rules are reversed.
- -a <minaqual>, --a=<minaqual>
Skip all reads with MAPQ alignment quality lower than the given minimum value (default: 10). MAPQ is the 5th column of a SAM/BAM file and its usage depends on the software used to map the reads.
- -t <feature type>, --type=<feature type>
Feature type (3rd column in GTF file) to be used, all features of other type are ignored (default, suitable for RNA-Seq analysis using an Ensembl GTF file:
exon)
- -i <id attribute>, --idattr=<id attribute>
GTF attribute to be used as feature ID. Several GTF lines with the same feature ID will be considered as parts of the same feature. The feature ID is used to identity the counts in the output table. The default, suitable for RNA-Seq analysis using an Ensembl GTF file, is
gene_id.
- --additional-attr=<id attributes>
Additional feature attributes, which will be printed as an additional column after the primary attribute column but before the counts column(s). The default is none, a suitable value to get gene names using an Ensembl GTF file is
gene_name. To use more than one additional attribute, repeat the option in the command line more than once, with a single attribute each time, e.g.--additional-attr=gene_name --additional_attr=exon_number.
- --add-chromosome-info
Store information about the chromosome of each feature as an additional attribute (e.g. column in the TSV output file).
- --feature-query=<query>
Restrict to features descibed in this expression. Currently supports a single kind of expression: attribute == “one attr” to restrict the GFF to a single gene or transcript, e.g. –feature-query ‘gene_name == “ACTB”’ - notice the single quotes around the argument of this option and the double quotes around the gene name. Broader queries might become available in the future.
- -m <mode>, --mode=<mode>
Mode to handle reads overlapping more than one feature. Possible values for <mode> are
union,intersection-strictandintersection-nonempty(default:union)
- --nonunique=<nonunique mode>
Mode to handle reads that align to or are assigned to more than one feature in the overlap <mode> of choice (see -m option). <nonunique mode> are
none,all,fraction, andrandom(default:none)
- --secondary-alignments=<mode>
Mode to handle secondary alignments (SAM flag 0x100). <mode> can be
scoreandignore(default:score)
- --supplementary-alignments=<mode>
Mode to handle supplementary/chimeric alignments (SAM flag 0x800). <mode> can be
scoreandignore(default:score)
- -o <samout>, --samout=<samout>
Write out all SAM alignment records into SAM files (one per input file needed), annotating each line with its feature assignment (as an optional field with tag ‘XF’)
- -c <countsoutput>, --counts_output=<countsoutput>
Filename to output the counts to instead of stdout. File format is autodetected based on the filename suffix (extension). Supported formats: tsv, csv, mtx, h5ad, loom. You need anndata for h5ad and loompy for loom support. For mtx, h5ad, and loom formats, the data type is float32.
- --counts-output-sparse
Store the counts as a sparse matrix. Only used for the following output file formats: mtx, h5ad, loom.
- -n <n>, --nprocesses=<n>
Number of parallel CPU processes to use (default: 1). This option is useful to process several input files at once. Each file will use only 1 CPU. It is possible, of course, to split a very large input SAM/BAM files into smaller chunks upstream to make use of this option.
- -p <samout_format>, --samout-format=<samout_format>
Format to use with the –samout option, can be
bamorsam(default:sam).
- -q, --quiet
Suppress progress report and warnings
- -h, --help
Show a usage summary and exit
- --version
Show software version and exit
Frequenctly asked questions
- My shell reports “command not found” when I try to run “htseq-count”. How can I launch the script?
The file “htseq-count” has to be in the system’s search path. By default, Python places it in its script directory, which you have to add to your search path. A maybe easier alternative is to write
python -m HTSeq.scripts.countinstead ofhtseq-count, followed by the options and arguments, which will launch the htseq-count script as well.
- Why are multi-mapping reads and reads overlapping multiple features discarded rather than counted for each feature?
The primary intended use case for
htseq-countis differential expression analysis, where one compares the expression of the same gene across samples and not the expression of different genes within a sample. Now, consider two genes, which share a stretch of common sequence such that for a read mapping to this stretch, the aligner cannot decide which of the two genes the read originated from and hence reports a multiple alignment. If we discard all such reads, we undercount the total output of the genes, but the ratio of expression strength (the “fold change”) between samples or experimental condition will still be correct, because we discard the same fratcion of reads in all samples. On the other hand, if we counted these reads for both genes, a subsequent diffential-expression analysis might find false positives: Even if only one of the gene changes increases its expression in reaction to treatment, the additional read caused by this would be counted for both genes, giving the wrong appearance that both genes reacted to the treatment.- I have used a GTF file generated by the Table Browser function of the UCSC Genome Browser, and most reads are counted as ambiguous. Why?
In these files, the
gene_idattribute incorrectly contains the same value as thetranscript_idattribute and hence a different value for each transcript of the same gene. Hence, if a read maps to an exon shared by several transcripts of the same gene, this will appear tohtseq-countas and overlap with several genes. Therefore, these GTF files cannot be used as is. Either correct the incorrectgene_idattributes with a suitable script, or use a GTF file from a different source.- Can I use htseq-count to count reads mapping to transcripts rather than genes?
In principle, you could instruct htseq-count to count for each of a gene’s transcript individually, by specifying
--idattr transcript_id. However, all reads mapping to exons shared by several transcripts will then be considered ambiguous. (See second question.) Counting them for each transcript that contains the exons would be possible but makes little sense for typical use cases. (See first question.) If you want to perform differential expression analysis on the level of individual transcripts, maybe ahve a look at our paper on DEXSeq for a discussion on why we prefer performing such analyses on the level of exons instead.
- For paired-end data, does htseq-count count reads or read pairs?
Read pairs. The script is designed to count “units of evidence” for gene expression. If both mates map to the same gene, this still only shows that one cDNA fragment originated from that gene. Hence, it should be counted only once.
- What happens if the two reads in a pair overlap two different features?
The same as if one read overlaps two features: The read or read pair is counted as ambiguous.
- What happend if the mate of an aligned read is not aligned?
For the default mode “union”, only the aligned read determines how the read pair is counted. For the other modes, see their description.
- Most of my RNA-Seq reads are counted as ``__no_feature``. What could have gone wrong?
Common causes include: - The
--strandedoption was set wrongly. Use a genome browser (e.g., IGV) to check. - The GTF file uses coordinates from another reference assembly as the SAM file. - The chromosome names differ between GTF and SAM file (e.g.,chr1in one file and jsut1in the other).- Which overlap mode should I use?
When I wrote
htseq-count, I was not sure which option is best and included three possibilities. Now, several years later, I have seen very few cases where the defaultunionwould not be appropriate and hence tend to recommend to just stick tounion.- I have a GTF file, how do I convert it to GFF?
htseq-count expects a GTF file so there’s no need to do that.
- I have a GFF file, not a GTF file. How can I use it to count RNA-Seq reads?
The GTF format specifies, inter alia, that exons are marked by the word
exonin the third column and that the gene ID is given in an attribute namedgene_id, and htseq-count expects these words to be used by default. If you GFF file uses a word other thanexonin its third column to mark lines describing exons, notifyhtseq-countusing the--typeoption. If the name of the attribute containing the gene ID for exon lines is notgene_id, use the--idattr. Often, its is, for example,Parent,GeneIDorID. Make sure it is the gene ID and not the exon ID.- How can I count overlaps with features other than genes/exons?
If you have GTF file listing your features, use it together with the
--typeand--idattroptions. If your feature intervals need to be computed, you are probably better off writing your own counting script (provided you have some knowledge of Python). Follow the tutorial in the other pages of this documentation to see how to use HTSeq for this.- How should I cite htseq-count in a publication?
Please cite HTSeq as follows: S Anders, T P Pyl, W Huber: HTSeq — A Python framework to work with high-throughput sequencing data. bioRxiv 2014. doi: 10.1101/002824. (This is a preprint currently under review. We will replace this with the reference to the final published version once available.)