Counting reads¶
A very typical use case for the HTSeq library is to for a given list of genomic features (such as genes,
exons, ChIP-Seq peaks, or the like), how many sequencing reads overlap each of the features. As a more
complex example for using HTSeq, we supply the script htseq-count, which takes a GTF file with
gene models and a SAM file and counts for each gene how many reads map to it; see Section htseq-count: counting reads within features.
The htseq-count script, however, has implementation details which were chosen with a specific use
case in mind, namely to quantify gene expression for subsequent testing for differential expression,
which is why, for example, the script does not count reads that map to multiple genes. For other
applications, different resolutions of such ambiguities might be desirable, and then, a bioinformatician
may want to create her own counting script. In the following, we expand on the coverage of this
topic in the Tour (A tour through HTSeq) and give building blocks which should make it possible to write such scripts
also for bioinformaticians with only modest knowledge of Python.
Preparing the feature array¶
Our general approach is to define a GenomicArrayOfSets and fill it with all the features we would
like to get counts for.
Similar to the code shown in the Tour, we prepare such an object from the GTF file for yeast as follows:
import HTSeq
gtf_file = HTSeq.GFF_Reader( "Saccharomyces_cerevisiae.SGD1.01.56.gtf.gz" )
exons = HTSeq.GenomicArrayOfSets( "auto", stranded=True )
for feature in gtf_file:
if feature.type == "exon":
exons[ feature.iv ] += feature.attr["gene_id"]
A few things might be noteworthy here: For each exon, we just store the gene ID in the genomic array. Hence,
all exons from the same gene are represented with the same string. This is deliberate, as we want to count
on the level of genes, not exons, but could be done differently: storing the whole feature object in
the GenomicArrayOfSets uses up noticeably more memory but allows to access more information in downstream
processing.
Also note that in a GTF file, an exon that appears in several transcripts appear once for each transcripts. Because all these exons are represented by the same name, they will be collapsed to a single value in the GenomicArrayOfSets.
GTF files are not the only source of feature annotations. One could, as well, read a BED file or other text file with genomic coordinates of, say, ChIP-Seq peaks, putative enhancers, or any other kind of data. For example, if we have a tab-separated text file with feature coordinates in four columns – feature ID, chromosome, start, end – we might use:
features = HTSeq.GenomicArrayOfSets( "auto", stranded=False )
for line in open( "myfeatures.txt" ):
fields = line.split( "\t" )
iv = HTSeq.GenomicInterval( fields[1], int(fields[2]), int(fields[3]) )
features[ iv ] += fields[0]
Here, we have assumed that the coordinates follow Python conventions: The first base of a chromosome is numbered 0, not 1, and the end position is not included in the interval. Remember to subtract or add 1 as necessary if this is not the case with your input data.
Counting ungapped single-end reads¶
We start with the easiest case, that of ungapped single-end reads. We first recapitulate points already shown in the Tour and then add further refinements in the following.
If we have a SAM file with unmapped reads, we might use the following code:
import collections
counts = collections.Counter( )
almnt_file = HTSeq.SAM_Reader( "my_alignments.sam" )
for almnt in almnt_file:
if not almnt.aligned:
count[ "_unmapped" ] += 1
continue
gene_ids = set()
for iv, val in features[ almnt.iv ].steps():
gene_ids |= val
if len(gene_ids) == 1:
gene_id = list(gene_ids)[0]
counts[ gene_id ] += 1
elif len(gene_ids) == 0:
counts[ "_no_feature" ] += 1
else:
counts[ "_ambiguous" ] += 1
for gene_id in counts:
print gene_id, counts[ gene_id ]
For the benefit of readers with only limited Python knowledge, we go through this code chunk step by step:
The variable counts contains a dictionary, which will associate gene IDs with read counts. We
use a variant of Python’s usual dict type, namely the Counter class from the collections
module in the standard library (from Python 2.7 onwards), which initialized any new key with
the value zero. (Users of Python 2.6 can use collections.defaultdict(int) instead.)
We then instantiate a SAM_Reader` object (If you have a BAM file, use BAM_Reader
instead) and run through all its record in a for loop. As described in the Tour, each record
in the SAM file is vprovided to the loop body in the variable almnt.
We first check whether the read might be unaligned, and if so, increment a special counter
that we call _unmapped (with an underscore as prefix to distinguish it from gene IDs).
For the aligned reads, the alignment’s genomic interval, almnt.iv, shows us the interval
covered by the read. Using this as an index to feature gives us a view on this
stretch of the feature container, in which we had stored the exons. The iterator
features[ almnt.iv ].steps() returns pairs of the form (iv, val), where iv is
a genomic interval and val is the set of gene IDs associated with the exons overlapping
this step. Using the |= operator, we get the union of the sets of all the steps in the
initially empty set gene_ids, which, at the end, contains the gene IDs of all genes
that the read overlaps. Remember that a set can contain each element at most once. Hence,
even if we see the same gene in several steps (for example because the read overlaps with
several exons), we still get it only once in gene_ids.
We then treat three possible cases, namely that the set gene_ids contains exactly one element,
that it is empty, or that it contains mroe than one element. The first case is the desired one:
The read overlaps with precisely one gene, and we hence increase the count for this gene by one.
Note that we need the idiom list(gene_ids)[0] to extract the name of this single gene from
the set. If the read did not overlap with a gene (len(gene_ids) == 0), we increase a special
counter that we call _no_feature.
What should we do if the read overlaps more than one gene? Here, one might now come up with
sophisticated logic to decide which gene to count the read for. To keep things simple, however,
we simply count the read for none of the overlapped genes and instead increase the
special counter _ambiguous .
In the final two lines, we loop through the counter to print out the counts.
Counting gapped single-end reads¶
CIGAR Operations¶
The above code can be used as is e.g. for ChIP-Seq data, but for RNA-Seq data, we need an additional ingredient: When sequencing RNA, many reads will pass over an exon-exon junction and hence align to two (or more) disjunct intervals on the genome, tyically with an intron in between. If the reads have been aligned with a splice-aware alignment tool, such gapped alignment is indicated in the SAM file by the CIGAR string.
HTSeq parses the CIGAR string and presents it in the cigar slot of a class:SAM_Alignment object
as a list of class:CigarOperation objects. As an example, consider a SAM alignment record
describing a read that has been aligned to position 1000 on the ‘+’’ strand of chromosome chr1,
with CIGAR string 20M300N30M2I8M. Following the SAM specification (please read it first if
you are unfamiliar with CIGAR strings), this means an alignment as depicted here:
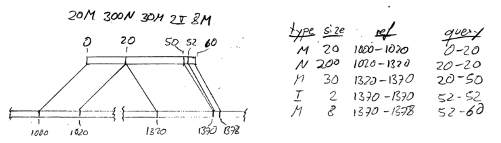
[[TO DO: Nicer image, add “chr1:”]]
The SAM_Alignment object will hence have in its cigar slot a list of 5 objects, each giving the
information of one row of the table. Note how some operations are associated with zero-length intervals
on either the reference (i.e., chromosome) or the query (i.e., read). For example, the intron (N200)
spans 200 bp on the chromosome (1020-1320) but a zero-length interval on the read (20-20). In this manner,
the CigarOperation class conveniently shows which intervals are affected by which operation.
Counting with gapped reads¶
In the code above, we used this for loop
gene_ids = set()
for iv, val in features[ almnt.iv ].steps():
gene_ids |= val
to collect the gene IDs of all exons overlapped by the reads interval. For loop runs over the whole interval covered by the aligned read, i.e., in the figure above, it would run from position 1000 to position 1378 on chromosome 1, including the alignment gap from 1020 to 1320. By looking at each cigar operation separately we can correctly skip the gaps. We only need to replace the for loop with the following double loop
gene_ids = set()
for cigop in almnt.cigar:
if cigop.type != "M":
continue
for iv, val in features[ cigop.ref_iv ].steps():
gene_ids |= val
The outer loop goes through the CIGAR operation, skipping all but the match operations, and
the inner loop inspects the steps covered by the match operations and collects the gene_ids in
the gene_ids set variable. The rest of the code stays as above.
Of course, custom logic can be implemented her to infer useful information from other cigar operation types, but for the simple counting task at hand here, we do not need this.
Dealing with multiple alignments¶
If the aligner finds multiple alignments for a read, these are typically reported in multiple
SAM records. If the SAM file unsorted or sorted by alignment position, it is hard to look at
all the possible alignments of a given read together, because the records with the alignments
for a given read are spread throughout the file. If the purpose of the counting is subsequent
testing for differential expression, it is often safest, anyway, to skip all multiply
aligned reads (because a read that is counted for several genes may cause spurious calls of
differential expression), and then, we merely need to recognize that a read has multiple
alignments. In the htseq-count script (see ref:htseqcount), this is done by two means:
First, many (but not all aligners) use the optional field “NH”, which indicates the number
of reported alignments. Testing for almnt.optional_field("NH") > 1 allows to find these
read. Also, if there are multiple good alignments, without one of them being considered by the
aligner to be more likely than the others, then the alignment quality (also denoted mapping quality,
MAPQ, in the SAM specification) should be 3 or less. Hence, if one skips all reads with an
alignment quality below, say, 10 (almnt.aQual < 10), one will skip over all multiply aligned
reads (provided the aligner indicates the mapping quality correctly, which is not always the case).
For more advanced use cases, it may be desirable to inspect all reported alignment, for example, to
the chose one using some custom logic, or to aggregate information over all of them. If the SAM
or BAM file has been sorted by read name then alternative alignments for the same read will
be in adjacent lines or records. To facilitate handling this case, HTSeq offers the function
function:bundle_multiple_alignments. It takes an iterator over SAM_Alignment objects
(e.g., a SAM_Reader or BAM_Reader object) and returns an iterator over
lists of SAM_Alignment objects. Each list contains only records describing alignments
for the same read. For this to work, the SAM file has to be sorted by read name to ensure that
mutiple alignments for the same read appear in adjacent records.
Handling paired-end reads¶
In the case of paired-end alignments, we will typically want to count read pairs, not reads. After all, the fragment (and not the reads from either of its ends) are the actual evidence for a gene’s expression that we want to count. Therefore, we want to process the alignment information for the two mated ends together.
First a quick review of how alignments for paired-end data are presented in a SAM file: The two “mated” reads referring to
the two end of a DNA fragment are reported in two separate records. The fact that the records describe
the same fragment can be seen from the fact that they have the same read name (given by the read.name
slot). That they refer to opposite ends can be seen from the respective bits in the FLAG field, which
are exposed by the SAM_Alignment.pe_which slot of the SAM_Alignment class, which takes
the values first or second (or unknown if not specified in the flag field, or not_paired_end if
an alignment of a single-end read is represented) . If the read pair has multiple
alignments, each alignment is reported by a pair of SAM records. As corresponding records are not necessarily
in adjacent lines, they are “linked” by the mate position fields (called RNEXT and PNEXT in the SAM specification),
which are exposed by the slot SAM_Alignment.mate_pos, which contains a GenomicPosition
object. The two records describing the two halves of a given alignment can be recognized as being correspondent
from the fact that each record’s mate_pos attribute is equal to the starting position (given by``iv.start_as_pos``).
Note that all the SAM records for a given read pair may be spread throughout the file. Only if the file is sorted by read name can we expect them to be at adjacent places, and even then, the records for multiple alignments can be intermixed.
To facilitate handling paired-end alignments, HTSeq offers the function pair_SAM_alignments(). This function
expects an iterator over SAM records (typically, a SAM_Reader or BAM_Reader object) and returns
an iterator over pairs (i.e., tuples of length 2) of SAM_Alignment records, with the first element being the alignment of the
read from the first sequencing pass (i.e., from the 5’ end of the DNA fragment) and the second element the corresponding alignment
from the second pass (i.e., the 3’ read). The function expects the SAM file to be sorted by read name. It proceeds by reading
in consecutive records with the same read name and storing them in a list. Once it finds a record with a differing read name,
the function goes through the list, sorts its content into pairs of corresponding alignment records and yields these pairs. If the
function’s option bundle``[TODO: add description of "bundle" in alignment.rst, too] is
set to ``True, the function does not yield the pairs separately but instead yields a list of all alignment pairs for
the same read.
Using these features, we can modify our counting loop as follows for paired-end data:
almnt_file = HTSeq.SAM_Reader( "my_paired_alignments.sam" )
counts = collections.Counter( )
for bundle in HTSeq.pair_SAM_alignments( almnt_file, bundle=True ):
if len(bundle) != 1
continue # Skip multiple alignments
first_almnt, second_almnt = bundle[0] # extract pair
if not first_almnt.aligned and second_almnt.aligned:
count[ "_unmapped" ] += 1
continue
gene_ids = set()
for iv, val in features[ left_almnt.iv ].steps():
gene_ids |= val
for iv, val in features[ right_almnt.iv ].steps():
gene_ids |= val
if len(gene_ids) == 1:
gene_id = list(gene_ids)[0]
counts[ gene_id ] += 1
elif len(gene_ids) == 0:
counts[ "_no_feature" ] += 1
else:
counts[ "_ambiguous" ] += 1
for gene_id in counts:
print gene_id, counts[ gene_id ]
Note that here, we skip reads if only one of two mates are aligned. Of course, one could choose as well to count such a pair for the gene to which the aligned mate has been mapped.
The need to sort paired-end SAM files by read name can be an inconvenience, because many aligners output the SAM file
sorted by position. In many use case, we can expect that the two ends of the same read will align to positions close
to each other on the genome. Then, an alternative strategy to pair up alignment records is go through the SAM file,
which has been sorted by position, and keep a dictionary of alignment records whose partner record has not been found yet.
For each record, we check the dictionary for its partner (i.e., for a record with the same read name and matching
position information). If we find the partner, we remove it from the dictionary and yield both together as a pair. If the partner
is not in the dictionary, the record is stored in the dictionary to wait for its partner to come along. As long as
mated records are not too far from each other in the file, the dictionary will only contain a manageable number of records.
Only if reads are often very far from each other (e.g., because the file was not sorted by position), the dictionary
might become too large to fit into memory. HTSeq offers this manner of pairing up alignment records in the function
pair_SAM_alignment_with_buffer(), which can be used in the same manner as pair_SAM_alignment(), but
takes one optional additional argument, the maximum size of the buffer (by default, 3 million).